Rheumatologische Erkrankungen
Kollagenosen
Antiphospholipid-Syndrom
Antiphospholipidsyndrom (APS, Antiphospholipid-Antikörper-Syndrom): Autoimmunerkrankung mit erhöhter Gerinnungsneigung, die zu wiederkehrenden Thrombosen und Embolien führt, bei Schwangeren auch zu Fehlgeburten. Auslöser sind die im Blut nachweisbaren Antiphospholipid-Antikörper. Frauen erkranken fünfmal häufiger als Männer, oft im mittleren Lebensalter. Der Verlauf der Erkrankung ist variabel, angefangen von einer einmaligen geringfügigen Venenthrombose bis hin zu tödlichen Lungenembolien oder Schlaganfällen. Behandelt wird das APS vor allem mit gerinnungshemmenden Medikamenten. Unter einer gut eingestellten Therapie ist die Lebenserwartung annähernd normal.
Symptome und Leitbeschwerden
- Schwellung eines Beins mit ziehendem Schmerz (tiefe Beinvenenthrombose)
- Plötzliche Atemnot mit stechendem atemabhängigem Schmerz in der Brust (Lungenembolie)
- Plötzliche Lähmung einer Körperseite oder Sprachstörungen bei jungen Menschen (Schlaganfall)
- Baumartig verzweigte bläuliche Hautzeichnung, kleine Einblutungen unter den Finger- oder Zehennägeln
- Wiederholte Spontanaborte vor der 12. Schwangerschaftswoche oder Fehlgeburten in den letzten beiden Dritteln der Schwangerschaft.
Wann in die Arztpraxis
Sofort den Notdienst rufen bei
- plötzlicher Lähmung, Sprachstörungen, plötzlicher Atemnot mit stechenden Brustschmerzen.
Am gleichen Tag bei
- Schwellung eines Beines mit ziehenden Schmerzen.
Demnächst, wenn
- ein Kinderwunsch besteht und es zu einer oder mehreren Fehlgeburten gekommen ist
- bläuliche Gefäßzeichnungen auf der Haut auftreten.
Die Erkrankung
Beim Antiphospholipid-Syndrom handelt es sich um eine Autoimmunerkrankung. Das heißt, dass der Körper Antikörper gegen eigene Strukturen bildet. Im Fall des APS sind dies Antikörper gegen Phospholipide.
Noch unklar ist, warum diese Antikörper zur Bildung von Blutgerinnseln führen. Man vermutet, dass sie u. a. mit einem Eiweiß reagieren, das an der Blutgerinnung beteiligt ist. Wenn die Gerinnsel Blutgefäße verschließen, kommt es zu Thrombosen und Embolien. Ist eine Frau schwanger, können die Gerinnsel die Blutversorgung der Plazenta stören und so eine Fehlgeburt auslösen.
Ursachen und Formen
Auch die Frage, warum und wie sich die Antiphospholipid-Antikörper im Körper bilden, ist noch nicht geklärt. In manchen Fällen scheint eine andere Krankheit der Auslöser dafür zu sein, dann handelt es sich um ein sekundäres APS. Am häufigsten ist das beim systemischen Lupus erythematodes der Fall. Aber auch Patient*innen mit einer rheumatoiden Arthritis, Krebserkrankung oder einer Infektion wie Hepatitis B oder AIDS können ein sekundäres APS entwickeln. Manchmal lösen Medikamente die Antikörperbildung aus, Beispiele sind Östrogene und Chlopromazin.
Bei der Hälfte aller Patient*innen lässt sich keine zugrundeliegende Erkrankung finden. Dann spricht man von einem primären, idiopathischen APS.
Klinik
Thrombosen können beim APS sowohl in Venen als auch in Arterien auftreten. Am häufigsten sind Beinvenenthrombosen und Lungenembolien, seltener bilden sich Venenthrombosen in Armen, Nieren, Leber und den Augenvenen. Durch die Thrombosen werden die Organe in ihrer Funktion eingeschränkt, es kommt zum Beispiel zu Nieren- oder Leberversagen, im Fall des Auges zu Sehstörungen bis hin zur Blindheit.
Arterielle Thrombosen entstehen vor allem in den Arterien des Gehirns und führen zu Schlaganfällen, selten auch zu Krampfanfällen, Migräne oder Demenz.
Eine weitere schwerwiegende Folge der erhöhten Thromboseneigung ist die Verstopfung der Gefäße, die in der Schwangerschaft die Plazenta versorgen. Dadurch kommt es vermehrt zu Schwangerschaftskomplikationen wie Fehlgeburten, Frühgeburten und Präeklampsie.
Komplikationen
Eine besonders gefährliche Komplikation ist das katastrophale APS, kurz CAPS genannt. Es tritt bei etwa 1 % der APS-Patient*innen auf und ist lebensbedrohlich. Dabei entstehen innerhalb kürzester Zeit massenhaft Thrombosen in mehreren Organsystemen (vor allem Lunge, Herz und Niere). Die betroffenen Organe werden nicht mehr ausreichend mit Blut versorgt und arbeiten nicht mehr richtig. Weil sich an den falschen Stellen Blutgerinnsel bilden, erschöpft sich außerdem das Gerinnungssystem. Es hat nicht mehr genug Kapazität für die physiologische Blutgerinnung und es drohen innere Blutungen. Dies und das Multiorganversagen führen trotz Intensivtherapie in jedem dritten Fall zum Tod.
Diagnosesicherung
Wenn aufgrund von Thrombosen oder Schwangerschaftskomplikationen der Verdacht auf ein APS besteht, lässt die Ärzt*in im Labor verschiedene Blutgerinnungswerte und das Blutbild untersuchen. Verdächtig sind eine Thrombozytopenie (also wenige Blutplättchen) und, als Hinweis auf die gestörte Blutgerinnung, ein verminderter Quick-Wert und eine verlängerte PTT (partielle Thromboplastinzeit).
Beweisend für das APS ist der Nachweis mindestens eines der drei im Blut vorkommenden Antiphospholipid-Antikörper:
- Lupus-Antikoagulans
- Anti-Cardiolipin-Antikörper
- Anti-β2-Glykoprotein-I-Antikörper.
Weil deren Bestimmung störanfällig ist, gibt es für die Diagnose strenge Kriterien. So müssen die Antikörper im Abstand von zwölf Wochen mindestens zwei Mal positiv sein. Außerdem sollen verschiedene Testsysteme verwendet werden.
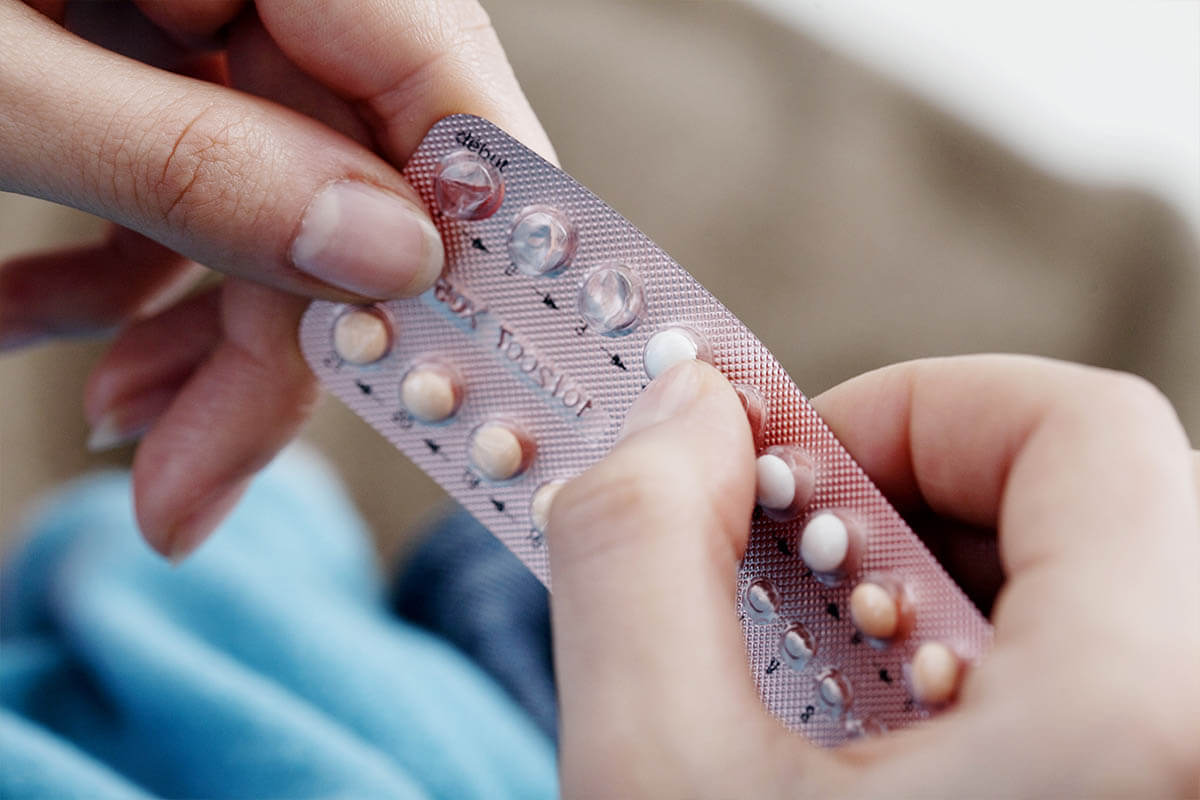
Differenzialdiagnose. Auszuschließen sind bei der Diagnose alle anderen Krankheitsbilder, die mit einer vermehrten Gerinnung verbunden sind. Dazu gehören der Mangel an Faktor II, Antithrombin III oder Protein S. Auch Krebs- und Nierenerkrankungen oder die Homocysteinämie können mit einer vermehrten Gerinnung einhergehen. Thrombosefördernd sind zudem Hormoneinnahmen (z. B. im Rahmen der Verhütung) oder das Rauchen.
Behandlung
Bei der Behandlung muss zwischen einem akuten Ereignis und der Vorbeugung von Thrombosen und Schwangerschaftskomplikationen unterschieden werden. Akute Beinvenenthrombosen, Schlaganfälle oder Herzinfarkte werden entsprechend therapiert (siehe dort).
Ob und welche gerinnungshemmende Therapie APS-Patient*innen vorbeugend benötigen, hängt nicht nur von den Antikörperwerten ab. Ein wichtiges Kriterium ist auch, ob schon eine arterielle oder venöse Thrombose stattgefunden hat oder ob es zu Schwangerschaftskomplikationen gekommen ist.
Primärprophylaxe. Bei manchen Patient*innen werden im Rahmen anderer Blutuntersuchungen zufällig Antiphospholipid-Antikörper gefunden, klinische Ereignisse wie Thrombose oder Fehlgeburt haben sie jedoch (noch) nicht erlebt. Ob diese Antikörperträger*innen eine vorbeugende gerinnungshemmende Therapie benötigen, ist umstritten. Wenn, dann verordnen die Ärzt*innen meist niedrig dosierte Acetylsalicylsäure (ASS). Zusätzlich hilft ein gesunder Lebensstil, um weitere thrombosefördernde Risikofaktoren zu reduzieren (siehe unten, Ihre Apotheke empfiehlt).
Sekundärprophylaxe. Nach jedem APS-bedingten Ereignis empfehlen die Leitlinien eine Sekundärprophylaxe mit einer gerinnungshemmenden Therapie. Sekundärprophylaxe bedeutet, nach einem stattgehabten Ereignis das nächste zu verhüten. Diese Vorbeugung wird an das Rezidivrisiko und die jeweilige Thrombose angepasst. Ein besonders hohes Risiko für ein erneutes Ereignis haben z. B. triple-positive Patient*innen. Das sind diejenigen, die alle drei Antiphospholipid-Antikörper im Blut aufweisen.
Nach einer Thrombose kommt meist eine dauerhafte Blutgerinnungshemmung mit einem Vitamin-K-Antagonisten (z. B. Phenprocoumon oder Warfarin) zum Einsatz. Ob der Gerinnungshemmer abgesetzt werden kann, wenn im Blut keine Antiphospholipid-Antikörper mehr nachweisbar sind, ist umstritten. Im Zweifel entscheidet die Ärzt*in dies gemeinsam mit der Patient*in nach dem individuellen Risikoprofil.
Im Spezialfall einer arteriellen Thrombose verordnen die Ärzt*innen häufig zusätzlich ASS.
Hinweis: Die zur Gerinnungshemmung ebenfalls eingesetzten direkten Antikoagulantien (DOAK, z. B. Dabigatran) sollen bei APS-Patient*innen nicht verwendet werden. Sie scheinen das Auftreten von thromboembolischen Ereignissen zu begünstigen.
Prophylaxe in der Schwangerschaft
Schwangere APS-Patientinnen haben ein hohes Risiko, Fehlgeburten oder eine Präeklampsie zu erleiden. Um diesen Schwangerschaftskomplikationen vorzubeugen, benötigen sie eine zuverlässige Blutgerinnungshemmung.
Vitamin-K-Antagonisten sind dafür nicht geeignet. Weil sie teratogen sind, also dem ungeborenen Kind schaden, dürfen sie weder kurz vor noch während einer Schwangerschaft verabreicht werden. Stattdessen empfiehlt man APS-Patientinnen mit Kinderwunsch zunächst ASS. Kommt es zu einer Schwangerschaft, erhält die werdende Mutter zusätzlich Heparin. Der Gerinnungshemmer muss bis zur sechsten Woche nach der Geburt täglich unter die Haut gespritzt werden.
APS-Expert*innen empfehlen ihren schwangeren Patientinnen meist, zusätzlich zur Vorbeugung mit Heparin auch ASS weiter einzunehmen. Allerdings nur über die ersten zwei Schwangerschaftsdrittel hinweg. Danach muss der Wirkstoff abgesetzt werden, weil er zum vorzeitigen Verschluss einer wichtigen Gefäßverbindung zwischen Aorta und Lungenschlagader des ungeborenen Kindes (Ductus botalli) führen kann.
Prognose
Unter Behandlung ist die Prognose gut und die Lebenserwartung des primären APS kaum verkürzt. Bei den sekundären Formen kommt es darauf an, welche Grunderkrankung vorliegt.
Ihre Apotheke empfiehlt
Was Sie selbst tun können
Gerinnungshemmer richtig einsetzen. Damit die Blutgerinnung zuverlässig gehemmt wird, müssen die Vitamin-K-Antagonisten täglich zur gleichen Zeit eingenommen werden. Auch die wöchentliche Kontrolle des Gerinnungswertes ist wichtig. Den sog. INR (International Normalized Ratio)-Wert kann man mit einem kleinen Messgerät und etwas Blut aus der Fingerbeere gut selbst bestimmen. Liegt er außerhalb des therapeutischen Bereichs, nimmt man Kontakt mit der Hausärzt*in auf, um die Dosierung gegebenenfalls anzupassen.
Ernährung beachten. Viele Nahrungsmittel enthalten Vitamin K und können deshalb die Wirkung von Vitamin-K-Antagonisten abschwächen. Dazu gehören z. B. Spinat, Brokkoli, Kichererbsen, Fenchel, Schnittlauch und Erbsen. Verzichten muss man deshalb auf das Gemüse nicht – man sollte jedoch jede Woche ungefähr die gleiche Menge davon zu sich nehmen. Bedacht werden muss zudem, dass die Wirkung der Vitamin-K-Antagonisten von Alkohol verstärkt und von Grapefruitsaft geschwächt werden kann.
Bewegen und Gewicht kontrollieren. Es gibt einige Maßnahmen, mit denen man selbst Thrombosen vorbeugen kann. Zur Sicherheit bespricht man sich jedoch vorher mit seiner Hausärzt*in, denn nicht alle sind für jede geeignet. Bewegung ist wichtig, um das Blut in Schwung zu bringen, günstig sind vor allem Radfahren, Schwimmen und Spazierengehen. Auch Treppensteigen statt Liftfahren freut die Beinvenen. Außerdem sollte man Übergewicht vermeiden bzw. reduzieren und ausreichend trinken. Wer im Beruf viel stehen muss, dem helfen oft Thrombosestrümpfe.
Mischkollagenose
Mischkollagenose (MCDT, Mixed Connective Tissue Disease, Sharp-Syndrom, Überlappungs-Syndrom, Overlap Syndrome, undifferenzierte Kollagenose): entzündliche Autoimmunerkrankung mit Merkmalen und Beschwerden
- eines systemischen Lupus erythematodes,
- einer Sklerodermie,
- einer Poly- oder Dermatomyositis und
- eines Sjögren-Syndroms.
Ein Teil der Patienten entwickelt im Verlauf das Vollbild einer der genannten Kollagenosen, meist einer Sklerodermie. Bei anderen Patienten bleibt langfristig das gemischte Beschwerdebild bestehen.
Polymyositis und Dermatomyositis
Polymyositis (PM): Seltene entzündliche Autoimmunerkrankung der Muskulatur. Sie zeigt sich vor allem an Oberarmen, Schultergürtel, Hüfte und Oberschenkeln mit fortschreitender Muskelschwäche und muskelkaterartigen Schmerzen. Oft kommt es zu Abgeschlagenheit und Fieber, zusätzlich können auch Herz, Lunge und Gelenke beteiligt sein.
Die Erkrankung beginnt meist zwischen dem 40. und 60. Lebensjahr und betrifft Frauen häufiger als Männer. Behandelt wird zunächst mit Kortison, im weiteren Verlauf kommen antirheumatische Wirkstoffe dazu. Die Prognose ist individuell sehr unterschiedlich und hängt auch davon ab, ob innere Organe betroffen sind.
Dermatomyositis (DM): Polymyositis mit Beteiligung der Haut an lichtexponierten Körperstellen. Die Hautveränderungen erscheinen oft lila, weshalb die Erkrankung auch Lilakrankheit genannt wird. Im Alter ist die Dermatomyositis häufig mit einer Tumorerkrankung assoziiert.
Symptome und Leitbeschwerden
- Muskelkaterartige Schmerzen
- Schwierigkeiten beim Aufstehen, Hinsetzen und beim Treppensteigen
- Probleme beim Armheben, Kämmen, Zähneputzen
- Müdigkeit, Abgeschlagenheit, Fieber, Gewichtsverlust
- Schwierigkeiten beim Schlucken.
Bei Dermatomyositis zusätzlich:
- Rötliche Schwellung der Oberlider
- Schuppende, entzündliche Hautrötungen, typischerweise von violetter Farbe, in Gesicht oder Ausschnitt
- Schuppende Hautrötungen über den Streckseiten der Fingergrund- und Mittelgelenke, an Ellenbogen, Knie und Knöchel (Gottron-Papeln)
- Hautverhärtungen, Pigmentstörungen und Schleimhautentzündungen.
Wann in die Arztpraxis
Demnächst, wenn
- oben genannte Beschwerden oder Hautveränderungen auftreten.
Die Erkrankung
Poly- und Dermatomyositis sind Autoimmunerkrankungen, bei denen der Körper aufgrund eines fehlgesteuerten Immunsystems körpereigene Strukturen angreift. Dadurch kommt es bei beiden Erkrankungen zur großflächigen Entzündung und dann zum Zerfall von Muskelfasern. Je nach Ausmaß sind weitere Gewebe wie Gelenke und innere Organe betroffen, bei der Dermatomyositis im Besonderen die Haut.
Die Ursache dieser Autoimmunkrankheiten ist unbekannt. Diskutiert werden eine genetische Veranlagung und verschiedene Viren. Bei manchen Betroffenen löst ein völlig anderer Mechanismus, nämlich Krebs (als paraneoplastische Symptome) die Krankheit aus. Bei der Dermatomyositis ist dies häufiger der Fall als bei der Polymyositis.
Klinik
Oft beginnen sowohl Poly- als auch Dermatomyositis schleichend mit unspezifischen Beschwerden wie Müdigkeit, Fieber oder Gewichtsverlust. Nach drei bis sechs Monaten machen sich zunehmend Muskelschmerzen bemerkbar, später auch Muskelschwäche und schließlich Lähmungen. Die Erkrankten haben z. B. Probleme, die Arme zu heben und sich die Haare zu kämmen. Oft fallen das Treppensteigen und das Aufstehen von einem Stuhl schwer. Bei Beteiligung der Speiseröhrenmuskulatur kommt es zu Schluckstörungen.
Bei der Dermatomyositis entwickeln sich um die Augenlider herum die typischen, lilafarbenen Hautveränderungen , auch heliotropes Erythem genannt. An den Streckseiten der Finger finden sich die ebenfalls typischen Gottron-Papeln. Dabei handelt es sich um lila-rötliche, flache Hautknötchen. Daneben gibt es zahlreiche weitere Hautveränderungen, wie z. B.
- Gebiete mit verstärkter oder abgeschwächter Pigmentierung
- derbe, verkalkte Hautareale
- erweiterte Hautgefäße
- flächige rote Erytheme um den Hals herum und im oberen Brustbereich
- pistolenholsterförmige rot-violette Bereiche am seitlichen Oberschenkel.
In unterschiedlichem Ausmaß sind auch die Gelenke und innere Organe von der Autoimmunerkrankung betroffen:
- Vor allem an Händen oder Knien sind die Gelenke geschwollen und schmerzen.
- Am Herzen kann es zu Herzschwäche, Rhythmusstörungen und Entzündungen der Herzkranzgefäße kommen.
- Bei jeder zehnten Patient*in ist auch die Lunge betroffen, meist in Form einer Lungenfibrose. Die Beschwerden sind trockener Husten und Luftnot.
Verlauf
Der Verlauf der Erkrankung lässt sich nicht vorhersagen. Bei manchen Patient*innen bessern sich die Beschwerden unter den Medikamenten oder die Krankheitsaktivität kommt immerhin zu einem Stillstand. Bei anderen schreitet die Myositis schubweise fort. Sind Herz oder Lunge beteiligt, führt häufig das Organversagen zum Tod.
Bei der Dermatomyositis sind Risikofaktoren für einen schweren Verlauf bekannt. Dazu gehören männliches Geschlecht, hohes Alter und eine Tumorerkrankung.
Komplikationen
Komplikationen ergeben sich z. B. dann, wenn die die Speiseröhre oder andere wichtige Organe betroffen sind. Schluckstörungen können so ausgeprägt sein, dass die Betroffenen mit einer Sonde ernährt werden müssen. Durch die muskuläre Schwäche der Speiseröhrensphinkter können Speiseteile in die Luftröhre und die Lunge gelangen (Aspiration). Befällt die Erkrankung die Atemmuskulatur, drohen Atemstörungen.
Eine Herzbeteiligung kann zu Herzversagen, die Lungenbeteiligung zum Versagen der Lungenfunktion führen.
Diagnosesicherung
Meist suchen die Betroffenen die Arztpraxis aufgrund der Muskelschwäche auf. Bei der klinischen Untersuchung stellt die Ärzt*in dann eine symmetrische Schwächung der Schulter und/oder Beckengürtelmuskulatur fest. Handelt es sich um eine Dermatomyositis, fallen die entsprechenden Hautveränderungen auf. Zum Nachweis der Diagnose dienen verschiedene Untersuchungsmethoden:
Blutuntersuchungen
Muskelenzyme. Durch den Zerfall der Muskulatur sind die Muskelenzyme im Blut erhöht, allen voran die Kreatinkinase. Weiterhin steigen im Blut die Werte von Myoglobin, Aspartat-Aminotransferase und Laktatdehydrogenase an.
Entzündungswerte. Als Zeichen der entzündlichen Vorgänge sind Blutsenkungsgeschwindigkeit und CRP erhöht, meist auch die Zahl der weißen Blutkörperchen (Leukozytose).
Autoantikörper. Etwa die Hälfte der Patient*innen weist erhöhte antinukleäre Antikörper (ANA) auf. Diese kommen allerdings auch bei anderen Autoimmunerkrankungen vor. Spezifisch für Polymyositis und Dermatomyositis sind dagegen Mi-2-Antikörper und Anti-Jo-1-Antikörper. Nachweisbar sind diese beiden Autoantikörper allerdings nur bei bis zu 30 % der Betroffenen.
Technische Verfahren
Bildgebung. Mit dem Ultraschall und der Kernspintomografie lassen sich Muskelschwellungen und Unregelmäßigkeiten der Muskelfasern gut erkennen.
Elektromyografie. Die Aktivität eines Muskels überprüft die Ärzt*in mithilfe der Elektromyografie (EMG). Betroffene Muskeln weisen darin eine veränderte elektrische Aktivität als Zeichen der Entzündung auf.
Muskelbiopsie. Entscheidend für die Diagnose ist die Muskelbiopsie. Darin finden sich abgestorbene Muskelfasern und eine hohe Anzahl von Immunzellen (T-Lymphozyten).
Spezialuntersuchungen zum Nachweis von Organmanifestationen. Mit Röntgen und Lungenfunktionsprüfungen weist die Ärzt*in eine Lungenbeteiligung nach. Bei Schluckproblemen kommen Ösophagus-Breischluck und Ösophagus-Manometrie zum Einsatz. Das Herz wird in der Regel mit EKG und Herzecho kontrolliert.
Tumorsuche
Weil die Polymyositis und im Besonderen die Dermatomyositis bei älteren Patient*innen oft mit Tumoren vergesellschaftet sind, muss die Ärzt*in bei bestätigter Diagnose nach einem bösartigen Tumor fahnden. Dazu gehören neben der gründlichen körperlichen Untersuchung u. a. das Röntgen von Lunge und Brustkorb, eine Koloskopie und die Ultraschalluntersuchung der inneren Organe. Bei Frauen kommen noch eine Mammografie und eine gynäkologische Untersuchung dazu.
Differenzialdiagnosen. Muskelschwäche und -schmerzen sind auch die Hauptbeschwerden bei zahlreichen anderen muskulären Erkrankungen. Dazu gehören die Polymyalgia rheumatica, die Myasthenie und medikamenteninduzierte Muskelerkrankungen, z. B. durch die Einnahme von Statinen. Eine weitere wichtige Differenzialdiagnose ist die Einschlusskörpermyositis.
Behandlung
Ziel der Behandlung ist die Verbesserung der Muskelkraft, die Verminderung der entzündlichen Vorgänge und die Vermeidung von Komplikationen.
Akuttherapie. Basis der medikamentösen Behandlung ist Kortison (z. B. Prednisolon), das zunächst hochdosiert verabreicht wird. Bessern sich die Beschwerden, reduziert die Ärzt*in die Kortisondosis schrittweise. Dabei orientiert man sich an den Muskelenzymen, die die Aktivität der Krankheit gut widerspiegeln. Ist die Erkrankung schon im Akutstadium schwer oder sind Organe beteiligt, kommen gleich Immunglobuline oder Immunsuppressiva zum Einsatz.
Langzeittherapie. Die Langzeittherapie besteht aus einer niedrig dosierten Kortisondosis und einem Immunsuppressivum (z. B. Azathioprin, Mycophenolatmofetil oder Methotrexat) für den Zeitraum von ein bis drei Jahren.
Spricht die Erkrankung auf die Behandlung nicht an, empfehlen die Leitlinien Wirkstoffe wie Rituximab, Tacrolimus (vor allem bei Lungenbeteiligung) und Cyclophosphamid. Eine weitere Therapieoption bei Therapieresistenz sind intravenös verabreichte, hoch dosierte Immunglobuline (IVIG).
Operation. Ist die Erkrankung mit einem Tumor assoziiert, führt häufig dessen Entfernung zu einer deutlichen Besserung der Beschwerden.
Behandlung von Schluckstörungen
Schluckstörungen sind bei Poly- und Dermatomyositis häufig und schränken die Lebensqualität der Betroffenen stark ein. Zur Behandlung gibt es verschiedene Möglichkeiten:
- Logopädie. Dabei stärkt man die Sprech- und Schluckmuskulatur mithilfe bestimmter Übungen.
- IVIG. Intravenös verabreichte Immunglobuline haben einen positiven Einfluss auf die Speiseröhrenmuskulatur.
- Botulinumtoxin-Injektionen. Mit der Injektion von Botulinumtoxin (Botox, bekannt zur Behandlung von Mimikfalten) lässt sich die Muskulatur der Speiseröhre entspannen und das Schlucken erleichtern.
- Myotomie und Ballondilatation. Bei diesen Verfahren wird die Speiseröhre von innen durch einen Schlauch behandelt (Endoskopie). Mithilfe der Ballondilatation dehnt man sie vorsichtig auf, bei der Myotomie durchtrennt die Chirurg*in gezielt bestimmte Muskelschichten.
Lassen sich die Schluckstörungen mit keiner der Methoden beherrschen, legen die Ärzt*innen eine Magensonde durch die Bauchwand (PEG-Sonde).
Physiotherapie
In der Akutphase ist körperliche Anstrengung verboten, häufig ist sogar Bettruhe erforderlich. Um die Gelenke beweglich zu halten, werden zunächst nur passive Übungen durch die Krankengymnast*in durchgeführt. Ist die Krankheitsaktivität unter Kontrolle, stehen aktive Übungen an. Sie dienen dazu, die Kraft der betroffenen Muskeln zu erhalten bzw. zu verbessern.
Prognose
Der Verlauf der Polymyositis ist sehr variabel, die Erkrankung kann unter Therapie sowohl zum Stillstand kommen als auch weiter fortschreiten.
Bei der Dermatomyositis ist die Prognose eher schlecht. Etwa ein Viertel der Betroffenen leidet langfristig unter Muskelschwäche und einer daraus folgenden Behinderung. In den ersten zwei Jahren nach Diagnose versterben etwa 30 % der Patient*innen, vor allem an den häufig zugrundeliegenden Tumorerkrankungen.
Ihre Apotheke empfiehlt
Was Sie selbst tun können
Körperlich schonen. In der akuten Phase der Erkrankung belastet körperliche Aktivität die Muskulatur zusätzlich. Deshalb sollte man sich während akuter Schübe schonen und eine verordnete Bettruhe unbedingt einhalten.
Muskelkraft trainieren. Außerhalb der akuten Phasen gilt es, die verbleibende Muskelkraft zu erhalten oder sogar zu verbessern. Mithilfe der Physiotherapie sollten entsprechende Übungen erlernt und regelmäßig durchgeführt werden.
Kontrolluntersuchungen wahrnehmen. Bei chronisch-entzündlichen Autoimmunerkrankungen ist es wichtig, dass die Ärzt*in die Medikamente regelmäßig an die Krankheitsaktivität anpasst. Außerdem müssen bei einigen der verordneten Wirkstoffe immer wieder das Blutbild und andere Laborwerte kontrolliert werden.
Vor UV-Strahlung schützen. Sonnenlicht verschlimmert die Hautveränderungen bei Dermatomyositis. Deshalb sollten Betroffene starke Sonneneinstrahlung unbedingt meiden und sich im Freien durch entsprechende Kleidung und Sonnencreme vor UV-Strahlung schützen.
Weiterführende Informationen
Die Myositis-Gruppe der Deutschen Gesellschaft für Muskelkranke e.V. bietet auf ihrer Webseite Informationen, Veranstaltungstipps und Hilfe zur Selbsthilfe für Betroffene.
Sjögren-Syndrom
Sjögren-Syndrom (Sjögren-Erkrankung, Dacryo-Sialo-Adenopathia atrophicans, primäres Sicca-Syndrom): chronisch voranschreitende Autoimmunerkrankung aus der Gruppe der Kollagenosen, von der meist Frauen betroffen sind. Das Sjögren-Syndrom befällt vor allem Tränen- und Speicheldrüsen, kann sich aber im Verlauf auch auf Schleimhäute, Gefäße, innere Organe und das Nervensystem ausbreiten. Die Leitbeschwerde ist das Sicca-Syndrom, also trockene Augen und ein trockener Mund. Zusätzlich schränken Erschöpfung, Depression und Leistungsminderung die Lebensqualität der Betroffenen erheblich ein. Das Sjögren-Syndrom ist nicht heilbar, mit den passenden Maßnahmen lassen sich die Beschwerden jedoch lindern. Sind innere Organe beteiligt, kommen meist immunsupprimierende Medikamente zum Einsatz.
Hinweis: Das Sjögren-Syndrom wird in zwei Formen eingeteilt: Beim (selteneren) primären Sjögren-Syndrom handelt es sich um eine eigenständige Erkrankung. Das sekundäre Sjögren-Syndrom entwickelt sich im Zusammenhang mit einer anderen Autoimmunerkrankung, wobei die Sicca-Problematik schon lange vor deren Diagnose auftreten kann.
Symptome und Leitbeschwerden
- Trockene, tränende und juckende Augen, Fremdkörpergefühl im Auge
- Trockener Mund, Schwierigkeiten beim Schlucken, Kekse essen unmöglich
- Nächtlicher Trinkzwang durch Mundtrockenheit
- Unerklärliches Fieber, starke Müdigkeit, Abgeschlagenheit
- Schwellung der Ohrspeicheldrüse
- Trockene Nase mit Borkenbildung, trockener Husten
- Schwellung der Halslymphknoten
- Gelenk- und Muskelschmerzen.
Wann in die Arztpraxis
In den nächsten Tagen, bei
- trockenen, geröteten und tränenden Augen ohne plausible Ursache, wie z. B. starkem Wind.
In den nächsten Wochen, wenn
- der Mund ungewöhnlich trocken ist.
Die Erkrankung
Vorkommen und Häufigkeit
In Deutschland leiden etwa 400 von 100.000 Einwohnern an einem primären oder sekundären Sjögren-Syndrom. Damit ist es nach der rheumatoiden Arthritis die zweithäufigste entzündlich-rheumatische Erkrankung. Zu 90 % sind Frauen betroffen, das mittlere Alter bei Diagnosestellung beträgt 56 Jahre. Allerdings gehen die Expert*innen davon aus, dass die Krankheit häufig viel früher beginnt und erst nach Jahren erkannt wird.
Am primären Sjögren-Syndrom sollen weltweit etwa 60 von 100.000 Menschen erkranken, verlässliche Daten für Deutschland gibt es nicht. Das sekundäre Sjögren-Syndrom ist häufiger, es tritt z. B. zusammen mit folgenden Autoimmunerkrankungen auf:
- Systemischer Lupus erythematodes (36 %)
- Rheumatoide Arthritis (32 %)
- Systemische Sklerodermie (24 %)
- Primär biliäre Zirrhose (50 %).
Krankheitsentstehung und Ursachen
Beim Sjögren-Syndrom wandern Zellen des Immunsystems, die Lymphozyten, in die exokrinen Drüsen ein. Exokrine Drüsen sind Drüsen, die Sekrete nach außen abgeben, also an eine innere oder äußere Körperoberfläche. Dazu gehören z. B. Tränen- und Speicheldrüsen, aber auch die Talg- und Schweißdrüsen und die Drüsen innerer Organe (Bronchialdrüsen, Prostatadrüsen).
In den Drüsen verdrängen und zerstören die Lymphozyten das gesunde Gewebe, sodass die Drüsen immer weniger Sekrete produzieren. Gleichzeitig werden bestimmte Immunzellen, die B-Lymphozyten, übermäßig aktiviert. Sie bilden dann spezielle Antikörper, die die Funktion der Drüsen zusätzlich verschlechtern. Diese Antikörper werden auch diagnostisch genutzt (siehe unten).
Diese autoimmun-entzündlichen Vorgänge bleiben jedoch nicht auf die Drüsen beschränkt. Sie breiten sich immer weiter aus und können auch innere Organe und das Nervensystem beeinträchtigen (siehe Organkomplikationen). Durch die krankhafte Aktivierung der Lymphozyten kann sich in seltenen Fällen langfristig auch ein Lymphom (Lymphdrüsentumor) entwickeln.
Ursachen. Beim sekundären Sjögren-Syndrom ist die zugrundeliegende Erkrankung der Auslöser für die Autoimmunreaktion. Die Ursache der primären Form ist noch unklar. Man weiß jedoch, dass hormonelle Faktoren, genetische Veranlagung, Umwelteinflüsse und verschiedene Immunmechanismen eine Rolle bei der Krankheitsentstehung spielen. Neuere Untersuchungen deuten darauf hin, dass womöglich virale Infektionen, vor allem über den Darm, als Trigger beteiligt sind.
Klinik und Verlauf
Als erstes macht sich die Erkrankung meist durch das Sicca-Syndrom bemerkbar. Die ausgeprägte Beeinträchtigung der Tränendrüsen führt zu trockenen Augen und Juckreiz, es drohen chronische Bindehautentzündungen. Der Mangel an Speichel begünstigt Karies, Parodontitis und Mundgeruch. Außerdem fällt den Patient*innen das Kauen und Schlucken schwer. Beim Befall der Ohrspeicheldrüsen schwellen diese an, und zwar meist auf beiden Seiten.
Weil auch die Nasenschleimhaut austrocknet, sind der Geruchs- und Geschmackssinn vermindert. Zudem drohen häufige Infektionen im gesamten Rachenraum. Auch die Schleimhaut der Vagina kann durch das Sicca-Syndrom austrocknen und deshalb jucken und schmerzen. Betroffene Frauen haben dadurch Schmerzen beim Geschlechtsverkehr und sind anfällig für Pilzinfektionen.
Sind die Schweißdrüsen betroffen, wird die Haut schuppig, trocknet aus und juckt.
Schon früh führt das Sjögren-Syndrom zu Erschöpfungszuständen (Fatigue) und Leistungsminderung. Viele Betroffene haben Probleme, sich zu konzentrieren. Fast ein Drittel leidet unter Depressionen.
Organkomplikationen
Bei etwa 30 % der Sjögren-Patient*innen entwickelt sich eine Arthritis mit Gelenkschmerzen und Funktionseinschränkungen. Bis zu 40 % haben Durchblutungsstörungen an den Fingern, die als Raynaud-Syndrom in Erscheinung treten. Entzündungen der kleinen und mittleren Gefäße verursachen Hauteinblutungen (vor allem an den Beinen) und Hautgeschwüre.
Jede vierte Patient*in bekommt neurologische Probleme wie Lähmungen oder Gefühlsstörungen. Jede Fünfte muss mit entzündlichen Veränderungen des Lungenzwischengewebes rechnen. Eine solche interstitielle Pneumonie kann zur Vernarbung (Fibrose) der Lunge führen, wodurch Husten, Luftnot und Kurzatmigkeit drohen.
Weitere, aber seltenere Ziele der Autoimmunkrankheit sind der Magen-Darm-Trakt (dann kommt es z. B. zur chronischen Magenschleimhautentzündung), die Niere und die Leber.
Lymphomrisiko
Beim primären Sjögren-Syndrom ist das Risiko für bösartige Lymphome deutlich erhöht. Hochrisikopatient*innen benötigen deshalb engmaschige Röntgen- und Ultraschallkontrollen. Zu dieser Gruppe gehören Patient*innen mit mindestens einem der folgenden Befunde:
- Dauerhafte Schwellung der Ohrspeicheldrüsen
- Kryoglobulinämie
- Lymphknotenschwellungen
- Keimzentren im Speicheldrüsengewebe
- Entzündete Hautgefäße (Vaskulitis).
Diagnosesicherung
Gefühlte Trockenheit in Mund und Auge sind zwar typisch für ein Sjögren-Syndrom, kommen aber auch bei vielen anderen Erkrankungen vor. Für eine sichere Diagnose müssen noch weitere Kriterien erfüllt sein.
Zunächst wird in einer gründlichen Befragung geklärt, wie ausgeprägt die Beschwerden sind. Für ein Sicca-Syndrom spricht z. B. eine tägliche beeinträchtigende Augen- oder Mundtrockenheit seit mehr als drei Monaten oder ein bleibendes Fremdkörpergefühl im Auge. Kann die Patient*in feste Speisen ohne zusätzliche Flüssigkeitszufuhr nicht mehr schlucken, ist dies ebenfalls ein deutlicher Hinweis. Objektiviert werden Mund- und Augentrockenheit dann meist mit verschiedenen Tests:
- Der Schirmer-Test dient der Messung der Tränenproduktion. Dazu hängt die Ärzt*in einen kleinen Filterpapierstreifen mit Millimeter-Einteilung in den Bindehautsack. Nach fünf Minuten liest man ab, wieviel Strecke davon benetzt wurde. Liegt diese unter 5 mm, handelt es sich um ein krankhaft trockenes Auge.
- Mit der Sialometrie misst man die Speichelmenge. Dazu sammelt die Patient*in ihren Speichel über zwei Minuten im Mundboden und lässt ihn in einen Sammeltrichter ablaufen. Als auffällig gilt ein Speichelfluss von weniger als 0,1 ml/min.
- Eine weitere Methode zur Messung der Speichelproduktion ist der Saxon-Test. Hierbei kaut die Patient*in zwei Minuten lang auf einer Mullkompresse, die davor und danach gewogen wird. Liegt der Gewichtsunterschied unter 2,75 g, ist dies ein Hinweis auf eine zu geringe Speichelproduktion.
Sprechen Krankengeschichte und die Ergebnisse der Funktionstest für eine Sicca-Problematik, muss noch das Sjögren-typische Autoimmungeschehen nachgewiesen werden. Dafür gibt es folgende Verfahren:
Blutuntersuchung. Zum Nachweis der Erkrankung im Blut ist die Bestimmung der spezifischen SS-B- und SS-A-Antikörper am aussagekräftigsten. Sie liegen bei etwa 70 % der Sjögren-Patient*innen vor. Ebenfalls oft erhöht sind der Rheumafaktor und die Antinukleären Antikörper. Diese Werte sind aber nicht so aussagekräftig wie die spezifischen SS-B- und SS-A-Antikörper.
Speicheldrüsenbiopsie. Um die Veränderungen im Drüsengewebe nachzuweisen, betäubt die Ärzt*in die Innenseite der Unterlippe und entnimmt mehrere kleine, stecknadelkopfgroße Speicheldrüsen (Lippenbiopsie). Diese Probe wird dann in der Pathologie untersucht. In manchen Kliniken führt man auch statt der Lippenbiopsie eine Biopsie der Ohrspeicheldrüse durch, vor allem, wenn diese dauerhaft angeschwollen ist. Dies dient nicht nur zur Diagnose eines Sjögren-Syndroms, sondern auch dem Ausschluss oder Nachweis eines Lymphoms (Lymphdrüsenkrebs).
Für die sichere Diagnose betrachtet man alle Befunde (Krankengeschichte, Tests, Antikörper und Biopsieergebnisse) gemeinsam und bepunktet diese. Erreicht die Patient*in einen bestimmten Wert, ist die Diagnose Sjögren-Syndrom wahrscheinlich.
Differenzialdiagnosen. Eine Sicca-Problematik findet sich bei sehr vielen Zuständen oder Erkrankungen. So lösen zahlreiche Medikamente Trockenheit im Mund und im Auge aus, z. B. Antidepressiva, Antihistaminika, Medikamente gegen Herzrhythmusstörungen, Entwässerungsmittel und viele mehr. Auch eine vorangegangene Strahlentherapie kann zu einem Sicca-Syndrom führen. Auszuschließen sind zudem Hornhautentzündungen und eine alters- oder menopausenbedingte Augentrockenheit. Wichtige Differenzialdiagnosen sind auch Fibromyalgie, Depression und systemische Erkrankungen wie Sarkoidose und Amyloidose.
Therapie
Die langsame Zerstörung der Drüsen lässt sich nicht aufhalten, auch nicht mit Therapien, die das Immunsystem unterdrücken. Trotzdem kann man die Beschwerden der Erkrankung mit den geeigneten Maßnahmen lindern. Dazu gehören nicht nur ärztlich verordnete Medikamente, sondern auch viele allgemeine Anpassungen des Lebensstils in Form der Selbsthilfe (siehe unten, Ihre Apotheke empfiehlt).
- Augentrockenheit. Gegen die Trockenheit der Augen helfen künstliche Tränen, tagsüber in Form von Tropfen, nachts als Augengele. Sie sollten frei von Konservierungsstoffen sein, da diese Tränenfilm und Augenoberfläche angreifen. Bei Hornhautschäden sollten nächtliche Augenverbände und Salben (z. B. Borsalbe) aufgelegt werden. Ciclosporin-Augentropfen haben eine antientzündliche Wirkung, meist werden sie als 0,1%ige Emulsion verordnet. Neuere Optionen sind das Verschließen des Tränenpünktchens mit einer Art Stöpsel (sog. Punktum Plugs), damit die vorhandene Tränenflüssigkeit langsamer abgeleitet wird. Auch extra große Kontaktlinsen mit Wasserspeicherfunktion können die Beschwerden lindern.
- Mundtrockenheit und Speichelmangel. Speichel lässt sich weniger gut ersetzen: Hier hilft vor allem häufiges Trinken in kleinen Schlucken, bei manchen Patient*innen auch künstlicher Speichel. In schweren Fällen versucht die Ärzt*in, mit Pilocarpin die Drüsenfunktion anzuregen. Die häufig als Begleiterscheinung der Mundtrockenheit vorliegenden Pilzinfektionen der Mundhöhle werden mit Nystatin behandelt.
- Trockene Nasenschleimhaut. Zum Befeuchten der Nasenschleimhaut eignet sich Nasenöl. Abschwellende Nasentropfen dürfen nicht eingesetzt werden, da sie die Trockenheit zusätzlich verstärken.
- Scheidentrockenheit. Hier helfen Scheidengele oder, nach der Menopause, östrogenhaltige Salben und Ovula, die Schleimhaut anzufeuchten und aufzubauen.
Organkomplikationen werden spezifisch behandelt, die Palette der Medikamente ist dementsprechend breit. Bei Gelenkbeschwerden kommen beispielsweise Kortison (auch als Gelenkspritze), nicht-steroidale Antirheumatika und Hydroxychloroquin zum Einsatz. Eine interstitielle Pneumonie erfordert Kortison, manchmal auch Cyclophosphamid oder Nintedanib. Bei Nervenschmerzen (Neuropathie) helfen bestimmte Antidepressiva oder Gabapentin, eventuell auch Kortison oder intravenös verabreichte Immunglobuline.
Prognose
Die Erkrankung ist zwar nicht heilbar, schreitet aber in der Regel nur langsam fort und hat insgesamt eine gute Prognose. Liegen keine schweren Organkomplikationen vor, entspricht die Lebenserwartung von Patient*innen mit primärem Sjögren-Syndrom dem der Allgemeinbevölkerung. 5 bis 10 % von ihnen entwickeln allerdings im Rahmen ihrer Erkrankung einen Lymphdrüsentumor. Wird dieser nicht frühzeitig erkannt und behandelt, ist die Prognose schlechter.
Beim sekundären Sjögren-Syndrom hängen Prognose und Lebenserwartung von der Grunderkrankung ab.
Ihre Apotheke empfiehlt
Schleimhäute befeuchten. Am wichtigsten ist es, ausreichend zu trinken, mindestens 2 Liter pro Tag! Versuchen Sie außerdem die Speichelproduktion mit kleinen Schlucken Zitronenwasser, (zuckerfreien) Kaugummis oder auch Lutschern am Laufen zu halten. Kauen Sie Ihre Mahlzeiten gut durch. In Innenräumen ist die Zimmerluft ausreichend zu befeuchten, besonders während der Heizperiode.
Augenpflege. Vermeiden Sie Wind, Wärmestrahlung, verrauchte Räume und Klimaanlagen, sie trocknen die Augen aus. Verwenden Sie nachts Augensalben oder -gele anstatt künstlicher Tränen, sie wirken deutlich länger und sorgen für einen ruhigeren Schlaf. Manche Betroffene profitieren nachts auch von Kühlkompressen. Tragen Sie im Sommer und an windigen Tagen eine Sonnenbrille, eventuell mit Seitenschutz (Fahrradbrille). Gehen Sie alle 3–6 Monate zur Augenärzt*in und besprechen Sie mit ihr die Augenprobleme.
Zahnpflege. Unerlässlich ist eine regelmäßige und gründliche Zahnpflege: Sie fördert zum einen den Speichelfluss und schützt zum anderen vor Karies und Mundpilzinfektionen, die bei verminderter Speichelproduktion schneller auftreten. Lassen Sie sich von Ihrer Zahnärzt*in beraten, ob lokale Fluoride zur Kariesprophylaxe sinnvoll sind.
Hautpflege. Schützen Sie die Haut mit fetthaltigen Lotionen. Meiden Sie starke Sonneneinstrahlung, die die Haut zusätzlich austrocknet.
Körperliches Training. Viele Sjögren-Patient*innen leiden unter starker Erschöpfung und Leistungseinbußen. Dieser oft als bleierne Müdigkeit empfundene Zustand wird Fatigue genannt und kommt bei rheumatischen Erkrankungen und Kollagenosen sehr häufig vor. Ein geeignetes Mittel dagegen ist leichtes aerobes Ausdauertraining, z. B. Schwimmen, Radfahren, Spazierengehen oder Fitnesstraining im Sportstudio. Lassen Sie sich von Ihrer Ärzt*in beraten, wieviel Sie sich davon zumuten dürfen.
Medikamente. Wenn Sie Medikamente einnehmen, bringen Sie diese zu jedem Besuch in der Arztpraxis mit, auch zur Augenärzt*in, denn viele Arzneimittel (vor allem Psychopharmaka) verringern als Nebenwirkung Tränen- und Speichelproduktion.
Weiterführende Informationen
sjoegren-erkrankung.de: Website des Selbsthilfe Netzwerks Sjögren Syndrom, mit Erfahrungsberichten, Tipps von Betroffenen, Veranstaltungsankündigungen und Hinweisen zu Studien.
www.sjoegren-syndrom.de: Website der Sjögren-Syndrom-Selbsthilfegruppe Darmstadt mit vielen interessanten Informationen und aktuellen Nachrichten zum Thema.
Systemische Sklerose
Systemische Sklerose (Sklerodermie, Systemsklerose, SS, Progressive Systemsklerose PSS): Chronische autoimmunologische Bindegewebserkrankung aus der Gruppe der Kollagenosen. Es kommt zu Verdickung und Verhärtung der Haut, mit oder ohne Beteiligung innerer Organe (z. B. Speiseröhre, Lunge, Nieren oder Herz), Gelenke oder Muskeln. Über 90 % der Patient*innen leiden unter einem sekundären Raynaud-Syndrom mit Durchblutungsstörungen vor allem an den Fingern. Frauen erkranken etwa viermal häufiger als Männer. Der Krankheitsbeginn liegt meist zwischen dem 40. und 50. Lebensjahr. Eine Heilung ist nicht möglich, stattdessen steht die Linderung der Beschwerden und die Behandlung der jeweiligen Organschäden im Vordergrund. Zum Einsatz kommen physikalische Maßnahmen, Kälteprophylaxe, Rauchverbot und eine immunmodulierende, antientzündliche Therapie, z. B. mit Glukokortikoiden und Antirheumatika.
Hinweis: Unterschieden von der systemischen Sklerose wird die zirkumskripte Sklerodermie. Sie zeichnet sich durch umschriebene Verhärtungen (Sklerose) der Haut aus, ohne dass dabei innere Organe betroffen sind oder eine Raynaud-Symptomatik auftritt. Diese Erkrankung wird auch Morphea genannt und von der Hautärzt*in behandelt.
Symptome und Leitbeschwerden
Hautveränderungen:
- Verhärtungen der Haut an Händen, Füßen und/oder Körperstamm
- Kleine Geschwüre, nach Abheilung Narben an den Fingerspitzen
- Engegefühl der Haut, Gefühl als sei der Körper "eingemauert"
- Gesichtsveränderungen, Mund wird kleiner
- Schmerzlose Wasseransammlungen an Händen und Füßen
- Stippchenförmige, durch die Haut hindurch schimmernde Kalkspritzer.
Weitere Beschwerden:
- Bei Kälte oder Stress plötzliches Weißwerden einzelner oder mehrerer Finger (Raynaud-Symptomatik)
- Allgemeine Müdigkeit und unerklärliche Mattigkeit, Fieber, Gewichtsabnahme
- Trockener Mund, trockene Augen
- Schluckbeschwerden, Sodbrennen
- Atemnot
- Gelenksteifigkeit, Gelenkschmerzen.
Wann in die Arztpraxis
Demnächst, wenn
- eine oder mehrere der oben genannten Beschwerden auftreten.
Die Erkrankung
Ausgangspunkt der systemischen Sklerose ist ein fehlgeleitetes Immunsystem. Es erkennt körpereigene Strukturen als fremd und regt die weißen Blutkörperchen an, Antikörper gegen das eigene Gewebe (Autoantikörper) zu bilden. Die Autoantikörper verteilen sich über das Blut im ganzen Organismus und verursachen an Organen, Gelenken und in der Haut Entzündungen. Gleichzeitig produziert der Körper mehr Bindegewebsfasern (Kollagen). Die überproduzierten Fasern lagern sich als Knoten und Verhärtungen in der Haut an und in den inneren Organen ab. Diese Ansammlungen von Bindegewebe (Fibrose) kann die Funktion der Organe massiv einschränken (siehe unten).
Zusätzlich kommt es durch die autoimmunen Prozesse zu einer Schädigung der Innenschicht der Blutgefäße. Dadurch wird der Gefäßinnenraum enger, was im ganzen Körper die Durchblutung stören kann.
Weil das ganze Körpersystem von der Erkrankung betroffen ist, spricht man von einer systemischen Erkrankung.
Epidemiologie und Ursache
Die systemische Sklerose ist selten, es erkranken etwa 50 von 100.000 Menschen in Deutschland daran. Frauen sind deutlich häufiger betroffen als Männer (3–5 : 1).
Die Ursache ist unklar, eine genetische Veranlagung ist anzunehmen. Diskutiert werden zudem verschiedene Krankheitsauslöser, beispielsweise Chemikalien wie organische Lösungsmittel, Benzin, Quarzstaub oder Formaldehyd. In Frage kommen auch bestimmte Medikamente oder Drogen, zum Beispiel Bleomycin, L-Tryptophan, Methamphetamin, Kokain und Appetitzügler wie Fenfluramin.
Klinik
Die systemische Sklerose wird in zwei Formen eingeteilt: die häufigere limitierte Form und die diffuse Form. Limitierte und diffuse Form unterscheiden sich darin, wie ausgeprägt die Beschwerden sind und in welchem zeitlichen Verlauf sie erscheinen.
Bei beiden Formen sind Finger, Zehen sowie Nase und Ohren verhärtet und verdickt. Das Gesicht ist maskenhaft starr, die Mundöffnung verkleinert (Mikrostomie). Die Haut ist aufgequollen, aber zugleich straff und verhärtet, oft lagert sich Kalk ein. Besonders an den Fingern entwickeln sich durch die Veränderungen an Haut und Bindegewebe Kontrakturen (Sklerodaktylie). Das bedeutet, dass die Gelenke in einer Stellung fixiert sind. Außerdem leidet fast jede Patient*in unter einem sekundären Raynaud-Syndrom.
Bei der limitierten Form beschränken sich die Hautveränderungen fast immer auf die Akren (Finger, Zehen, Nase, Ohren) und das Gesicht. Innere Organe sind meist erst sehr spät betroffen.
Die seltenere diffuse Form ist weitaus aggressiver. Die Hautveränderungen breiten sich über den gesamten Körper und das Gesicht aus. Außerdem leiden die Patient*innen meist erheblich unter trockenen Schleimhäuten, was sich vor allem an den Augen und im Mund bemerkbar macht. Oft entzünden sich auch die Gelenke. Diese schmerzen dann und werden steif. Innere Organe sind bei der diffusen Form früh und ausgeprägt beteiligt:
- Bei etwa der Hälfte der Patient*innen ist die Speiseröhre betroffen: ihre Wand wird zunehmend starr, Schluckbeschwerden sind die Folge.
- Als zweithäufigstes Organ wird die Lunge in Mitleidenschaft gezogen, in Form einer Lungenfibrose mit trockenem Husten und Atemnot. Das zunehmende Bindegewebe versteift die Lunge und behindert das Einatmen.
- Am Herzen drohen Muskelentzündungen und in der Folge Herzschwäche und Herzrhythmusstörungen. Bei einer Beteiligung der Niere kann es zu einem Nierenversagen kommen.
Das CREST-Syndrom ist eine Sonderform der systemischen Sklerose, die vor allem Frauen über 50 Jahre betrifft. Durch die autoimmunbedingte Überproduktion von Kollagenfasern kommt es zu folgenden fünf Symptomen:
- Calcinosis cutis (knotige Kalkeinlagerungen in der Haut)
- Raynaud-Syndrom
- Bewegungsstörungen der Speiseröhre (englisch Speiseröhre = esophagus)
- Sklerodaktylie (gespannte und verhärtete Haut an den Fingern)
- Teleangiektasien (erweiterte Blutgefäße in der Haut, Besenreiser).
Zusätzlich können eine Lungenfibrose oder ein Bluthochdruck im Lungenkreislauf auftreten (pulmonale Hypertonie).
Diagnosesicherung
Eine Verdachtsdiagnose stellen Ärzt*innen bereits nach der klinischen Untersuchung, wenn ihnen die typischen Verhärtungen an Fingern und Zehen auffallen. Auch ein Raynaud-Syndrom spricht für eine Sklerodermie. Um sicher zu gehen, gibt es weitere Untersuchungen:
Kapillarmikroskopie des Nagelfalzes. Dafür tropft die Ärzt*in etwas Öl auf das Nagelbett (die Nagelfalz) und untersucht es unter einem herkömmlichen Mikroskop. Im Frühstadium sind die Kapillaren (kleinste Blutgefäße) erweitert und manchmal auch ausgesackt. Im Spätstadium ist die Anzahl der Kapillaren deutlich vermindert und es gibt gefäßfreie Zonen. Manchmal lassen sich auch vermehrt Pigmentablagerungen erkennen. Die Kapillarmikroskopie ist wichtig, um das sekundäre Raynaud-Syndrom im Rahmen einer systemischen Sklerose von einem primären Raynaud-Syndrom zu unterscheiden. Bei Letzterem zeigen sich keine Veränderungen der Nagelfalzkapillaren.
Hautbiopsie. Mit einer Hautbiopsie lässt sich die Diagnose bestätigen. Dazu entnimmt die Ärzt*in eine Hautprobe aus einer verhärteten und verdickten Stelle und untersucht sie feingeweblich unter dem Mikroskop. Typische Merkmale sind z. B. verbreiterte Faserbündel aus Bindegewebe und eine verringerte Anzahl von Gefäßen.
Laboruntersuchungen. Bei den Laboruntersuchungen sprechen vor allem die typischen Antikörper für eine systemische Sklerose. Allerdings kann die Erkrankung auch vorliegen, ohne dass diese erhöht sind. Am wichtigsten ist der Nachweis von antinukleären Antikörpern (ANA), die bei 90 % der Patient*innen vorhanden sind. Etwa ein Drittel der Betroffenen hat auch einen positiven Rheumafaktor. Folgende Antikörper haben ebenfalls diagnostische Bedeutung:
- Anti-Scl-70-Antikörper sind bei etwa 40 % der Patient*innen mit diffuser systemischen Sklerose positiv und sprechen für eine schlechte Prognose.
- Antizentromer-Antikörper finden sich vor allem bei der limitierten Form und beim CREST-Syndrom.
- Anti-RNA-Polymerase III-Antikörper sind mit einer prognostisch ungünstigen Nierenbeteiligung assoziiert.
Weitere Laboruntersuchungen helfen dabei, das Ausmaß der Erkrankung zu erkennen. Die Entzündungswerte (CRP, BSG) zeigen, wie stark die Entzündung ausgeprägt ist. Anhand von Kreatininwert und Eiweißausscheidung im Urin weist man eine Nierenbeteiligung nach. Eine Muskelentzündung sieht die Ärzt*in am Anstieg bestimmter Muskelenzyme im Blut.
Apparative Diagnostik. Bei Schluckbeschwerden zeigen Ösophagus-Breischluck und die Speiseröhrendruckmessung, ob die Speiseröhre beteiligt ist. Um eine Lungenfibrose frühzeitig zu erkennen, werden regelmäßig CT-Aufnahmen der Lunge angefertigt und die Funktion der Lunge untersucht. Je nachdem, welche Organe befallen sind, sind auch Ultraschalluntersuchungen, EKG, Herzechokardiografie und Kontrastmitteldarstellungen erforderlich.
Behandlung
Die Behandlung der systemischen Sklerose richtet sich nach den betroffenen Organen. Neben dem Erhalt bzw. der Verbesserung der Lebensqualität ist es wichtig, den Befall innerer Organe frühzeitig zu erkennen und ihre Zerstörung aufzuhalten.
Basis der Therapie sind allgemeine Maßnahmen (siehe Ihre Apotheke empfiehlt) und eine immunmodulierende und antientzündliche Therapie, die an das Ausmaß der systemischen Entzündung angepasst wird. Dazu kommt die an spezielle Komplikationen angepasste medikamentöse oder operative Behandlung.
Immunmodulatorische Therapie
Wenn allgemeine Maßnahmen (siehe unter Ihre Apotheke empfiehlt) zur Linderung der Hautproblematik nicht ausreichen oder wenn innere Organe beteiligt sind, verordnen die Ärzt*innen in der Regel eine antientzündliche bzw. immunmodulatorische Therapie. Dadurch sollen die Entzündung und der fibrotische Umbau von Haut und Organen aufgehalten werden.
- Medikamente. Neben Kortison werden verschiedene antirheumatische Wirkstoffe eingesetzt. Dazu gehören Cyclophosphamid, Azathioprin, Methotrexat, Mycophenolat-Mofetil oder Tocilizumab. Sie setzen an unterschiedlichen Stellen des Immunsystems an und bremsen dadurch die entzündlichen Vorgänge.
- Extrakorporale Photopherese. Bei dieser in speziellen Zentren durchgeführten Behandlung werden der Patient*in zunächst mithilfe einer Blutwäsche weiße Blutkörperchen (Immunzellen) entnommen, diese mit UV-Licht bestrahlt und wieder zurückinfundiert. Die veränderten, zurückinfundierten Blutkörperchen bewirken dann im Körper, dass die krankhafte Immunreaktion abgemildert wird.
- Plasmapherese. Sind die Antikörper-Titer im Blut sehr hoch, kann eine Plasmapherese helfen. Dabei handelt es sich um eine Art Blutwäsche, wodurch die Antikörper aus dem Blut entfernt und damit zumindest zeitweise reduziert werden.
- Stammzelltransplantation. Dieses sehr riskante und aufwändige Verfahren wird eingesetzt, wenn keine anderen Maßnahmen mehr helfen und innere Organe wie die Lunge schwer betroffen sind. Dadurch werden die kranken Immunzellen entfernt und durch gesunde Immunzellen eines Spenders ersetzt.
Spezielle Therapien
Gegen Gelenkschmerzen kommen nichtsteroidale Antirheumatika wie Diclofenac oder Ibuprofen zum Einsatz. Bei starken Gelenkschmerzen und -schwellungen hilft auch niedrig dosiertes Kortison. Lassen sich die Schmerzen so nicht beherrschen, verordnet die Ärzt*in auch Opioide.
Trockene Augen lindert man mit Dexpanthenol-Augentropfen oder künstlichen Tränen. Bei Mundtrockenheit empfehlen sich künstliche Speichelpräparate, Kaugummikauen und reichliches Trinken (siehe auch unter Ihre Apotheke empfiehlt).
Bei Raynaud-Symptomatik kann das Auftragen von nitrathaltiger Salbe die Durchblutung an den betroffenen Stellen verbessern. Nitratsalbe gibt es in Deutschland allerdings nicht als Fertigpräparat, sie muss in der Apotheke angemischt werden. Sind die Beschwerden sehr stark, verordnet die Ärzt*in auch gefäßerweiternde Kalziumantagonisten in Tablettenform.
Zur Behandlung schwerer Durchblutungsstörungen dient die intravenöse Gabe von Prostaglandinpräparaten oder Iloprost. Sind Arterien betroffen, ist manchmal ein gefäßchirurgischer Eingriff erforderlich (z. B. das Legen einer Art Bypass bei Befall der A. radialis (Speichenarterie).
Kalkeinlagerungen in der Haut (Calcinosis cutis) erfordern ebenfalls ein chirurgisches Eingreifen. Oberflächliche Knoten schabt man aus (Kürettage), tiefere entfernt die Ärzt*in mit dem Skalpell oder dem Laser.
Zur Therapie der interstitiellen Lungenfibrose gibt es seit einigen Jahren vielversprechende Wirkstoffe. Behandelt wird zweigleisig, und zwar sowohl gegen die Entzündung (z. B. mit Cyclophosphamid, Tocilizumab und Mycophenolat-Mofetil) als auch gegen die Bindegewebsvermehrung (antifibrotisch). Zugelassen zur Therapie der Lungenfibrose bei systemischer Sklerose ist der Wirkstoff Nintedanib. Durch seine antifibrotische Wirkung kann Nintedanib den bindegewebigen Umbau der Lunge abmildern und die fortschreitende Atemnot lindern. Weitere Wirkstoffe sind in der klinischen Erprobung.
Prognose
Die Prognose der systemischen Sklerose ist individuell sehr unterschiedlich. Sie hängt maßgeblich vom Befall innerer Organe ab, wobei die Entwicklung einer Lungenfibrose den größten Einfluss auf die Lebenserwartung hat.
Mit einer adäquaten Behandlung kann das Fortschreiten oft verzögert oder zu einem Stillstand gebracht werden.
Ihre Apotheke empfiehlt
Bei der Behandlung der systemischen Sklerose sind folgende Basismaßnahmen in jedem Stadium essenziell:
Rauchen abgewöhnen. Nikotin verengt die Blutgefäße und vermindert die Durchblutung. Die durch die systemische Sklerose ohnehin verschlechterte Blutversorgung von Akren und Organen wird dadurch weiter vermindert. Sklerosepatient*innen sollten deshalb auf jeglichen Nikotingenuss verzichten.
Kälte vermeiden. Beim Raynaud-Syndrom sollten die Hände immer und überall möglichst warm gehalten werden. Neben Handschuhen helfen Pulswärmer dabei. Auch sonst ist Kälte zu meiden: Das bedeutet z. B., die Wohnung gut zu heizen und auf Wintersport zu verzichten. Viele Betroffene empfinden Wärmepackungen, warme Bäder und Infrarot-Kabinen als hilfreich.
Hautpflege und Kleidung. Das zunehmende Spannen der Haut lässt sich durch ölhaltige Bäder, Duschcremes und regelmäßiges Eincremen der Haut mit fetthaltigen Lotionen etwas mildern. Gut passende, weiche Schuhe (hinten eng und vorne weit) und weite Kleidung helfen, schlecht heilende Druckstellen zu vermeiden.
Mikrostomie. Sehr belastend für Patient*innen mit systemischer Sklerose ist die Mikrostomie, d. h. das durch Haut- und Bindegewebsschrumpfung Kleinerwerden des Mundes. Dem muss frühzeitig mit logopädischen Dehnübungen vorgebeugt werden.
Essen und Trinken. Bei Schluckstörungen zum Essen viel trinken und gut kauen. Bei gleichzeitiger Refluxkrankheit nicht direkt nach dem Essen hinlegen, da dies vermehrt Magensäure in die Speiseröhre zurückfließen lässt. Das Essen sollte auf viele kleine Mahlzeiten verteilt und fette Speisen am Abend gemieden werden.
Gymnastik. Zur Erhaltung der Beweglichkeit tragen gymnastische Übungen bei, z. B. Dehnen nach dem Aufstehen und nach Wärmeanwendungen oder auch Gymnastik im warmen Wasser.
Zahnpflege. Bei trockenem Mund oder erschwerter Mundöffnung auf intensive Zahnpflege achten, da hier vermehrt Karies auftritt. Zwischendurch helfen zuckerfreie Kaugummis oder kleine Schlucke Wasser, am besten mit Zitronensaft.
Psychotherapie. Die systemische Sklerose mit all ihren Manifestationen ist eine sehr belastende Erkrankung. Betroffene sollten sich nicht scheuen, psychotherapeutische Unterstützung zu suchen. Das Erlernen und Anwenden von Entspannungsverfahren kann helfen, besser mit den Schmerzen und krankheitsbedingten Einschränkungen umzugehen.
Systemischer Lupus erythematodes
Systemischer Lupus erythematodes (SLE, Schmetterlingsflechte, Lupus visceralis, Lupus disseminatus): Chronische Autoimmunkrankheit mit Befall zahlreicher Organe und unterschiedlichsten Verlaufsformen. Die häufigsten Symptome sind unklare Fieberschübe, Gelenkbeschwerden und Hautveränderungen. Die ~ 35 000 Betroffenen in Deutschland sind zu 90 % weiblich, das bevorzugte Erkrankungsalter liegt um die 30 Jahre.
Da sowohl die Schwere des Befalls einzelner Organe als auch der Verlauf stark variieren, sprechen manche Ärzte davon, dass die Krankheit „würfelt“: chronisch schubförmige, seltener auch kontinuierlich fortschreitende Verläufe, der Wechsel der betroffenen Organe – alles ist möglich. Manchmal heilt der SLE sogar aus.
Medikamentös induzierter Lupus erythematodes: Durch Medikamente ausgelöste Sonderform, z. B. durch bestimmte Antibiotika, Antiepileptika, Antirheumatika, Psychopharmaka, Thyreostatika und Antihypertensiva. Die Symptome verschwinden nach Absetzen der Medikamente. Viele dieser Medikamente sind mittlerweile nicht mehr gebräuchlich, daher ist der medikamentös induzierte Lupus selten geworden.
Die Erkrankung
Beim Lupus erythematodes ist die Immunregulation gestört. Die Erkrankung bricht oft während oder nach einer Schwangerschaft aus, die Einnahme der Pille kann die Erkrankung fördern und nicht selten verschwindet sie nach der Menopause wieder. Äußere Faktoren wie UV-Strahlen (Sonnenbäder) sorgen nicht nur für Hautveränderungen, sondern können auch Schübe auslösen. Für eine genetische Veranlagung spricht die Häufung in unterschiedlichen Bevölkerungsgruppen, in Asien tritt die Erkrankung doppelt, in Afrika sieben- bis achtmal so häufig auf wie in Europa.
Fast immer fühlen sich die Patienten müde, krank und abgeschlagen oder haben Fieber. Dazu kommen weitere organspezifische Beschwerden und Befunde:
- Hauterscheinungen (insgesamt 90 %): an sonnenlichtexponierten Stellen Rötungen, Hornhautverdickungen und Pigmentstörungen. Bei 50 % der Patienten zeigt sich die typische schmetterlingsförmige Hautrötung im Gesicht (Schmetterlingserythem). 20 % haben ein sekundäres Raynaud-Syndrom. Aber auch Symptome wie kreisrunder, meist bestehen bleibender Haarausfall oder Mundschleimhautentzündungen treten auf. Manchmal befällt der Lupus erythematodes ausschließlich die Haut. Häufig sind dann nicht einmal im Blut die sonst typischen Autoantikörper zu finden (diskoider Lupus erythematodes, auch bekannt als Hautlupus und kutaner Lupus erythematodes).
- Gelenkprobleme (90 %): Meist morgens schmerzhafte, geschwollene Gelenke v. a. im Knie- und Handbereich, häufig sind die Sehnenscheiden mitbefallen. Die Gelenke werden nicht zerstört.
- Blutbildveränderungen mit Blutarmut (also Mangel an roten Blutkörperchen), aber auch Leuko- und/oder Thrombozytopenie (Mangel an weißen Blutkörperchen bzw. Blutplättchen).
- Rippenfellentzündungen (50 %) mit wiederkehrenden starken Schmerzen beim Atmen.
- Eine Glomerulonephritis (45 %) oder andere Entzündung der Niere (45 %).
- Herzbeutel-, gelegentlich auch Herzmuskel- oder Herzinnenhautentzündungen (40 %).
- Verschiedene Störungen des zentralen Nervensystems (30 %): Dazu zählen Krampfanfälle und Psychosen, starke Kopfschmerzen, die häufig auch einen neuen Schub ankündigen, Depressionen, Störungen von Merkfähigkeit und logischem Denken.
Das macht der Arzt
Diagnosesicherung. Da die Symptome stark variieren, ist eine sichere Diagnose in frühen Stadien nur zusammen mit Laborbefunden möglich. Im Blut nachgewiesen werden gering erhöhte Entzündungswerte sowie antinukleäre Antikörper (ANA) und deren Untergruppen (ds-DNS-AK). Um eine Nierenentzündung zu erkennen, wird der Urin auf Eiweiß untersucht (Proteinurie). Bei nachgewiesener Proteinurie hilft eine Nierenbiopsie, das Ausmaß der Schädigung zu beurteilen.
Therapie. Eine Heilung ist grundsätzlich nicht möglich, Ziel der Behandlung ist vielmehr, die Schübe zu verhindern oder wenigstens abzukürzen, die Beschwerden zu lindern sowie dauerhafte Organschäden zu vermeiden. Besonderes Augenmerk gilt dem Erhalt der Nierenfunktion. Aufgrund des kaum vorhersehbaren Verlaufs ist die Kontinuität der ärztlichen Betreuung und das Vertrauensverhältnis zwischen Rheumatologen und Patient besonders wichtig.
Die medikamentöse Therapie folgt dem Grundsatz, „so wenig wie möglich, aber so viel wie nötig“:
- Bei geringer Krankheitsaktivität (ohne Organbeteiligung) kommen viele Patienten lange Zeit mit nichtsteroidalen Antirheumatika wie Diclofenac (z. B. Voltaren®) aus. Die Antimalariamittel Chloroquin (Resochin®) oder Hydroxychloroquin (Quensyl®) und/oder Kortison sind bei ausgeprägten Hautveränderungen und Gelenkbeschwerden notwendig.
- Bei mittlerer Krankheitsaktivität (ohne Beteiligung von Herz, zentralem Nervensystem oder Nieren) wird der immunsuppressive Wirkstoff Azathioprin (Imurek®) eingesetzt.
- Wenn lebenswichtige Organe wie Nieren, Lunge, Gehirn oder Rückenmark bedroht sind, wird hoch dosiert Kortison sowie Cyclophosphamid (Endoxan®, ein relativ gut verträgliches Zytostatikum), eingesetzt, Letzteres häufig als Infusion. Reichen auch diese Medikamente nicht aus oder werden nicht vertragen, stehen weitere Zytostatika wie Methotrexat (Lantarel®), Cyclosporin (Sandimmun®), Mycophenolat oder hoch dosierte Immunglobuline zur Verfügung.
Selbsthilfe
Die Konsequenzen der Krankheit für die Betroffenen sind unterschiedlich. Die meisten Patienten sind im täglichen Leben wenig beeinträchtigt, für andere ist der Lupus so einschneidend, dass volle Berufstätigkeit nicht mehr möglich ist und im Haushalt Hilfe benötigt wird.
Um Verzweiflung und Resignation keine Chance zu geben, sind drei Dinge wichtig:
- Lernen Sie Ihre Krankheit verstehen. Ratgeber können helfen. Wenn eine Selbsthilfegruppe in der Nähe ist, nehmen Sie Kontakt auf.
- Finden Sie den Arzt Ihres Vertrauens und arbeiten Sie konsequent mit ihm zusammen. Das heißt vor allem, dass Sie die vereinbarten Medikamente einnehmen. Wenn Sie das nicht mehr in der bisherigen Dosierung wollen, besprechen Sie es vorher offen mit Ihrem Arzt. Nehmen Sie die Kontrolltermine wahr, gerade Nierenprobleme merken Sie selbst nicht. Sie riskieren sonst bleibende Schäden bis hin zu Organzerstörungen. Gehen Sie bei Verschlimmerungen, aber auch bei Beschwerden, die Sie nicht mit Ihrem Lupus in Verbindung bringen, zum Arzt. Die Symptome beim Lupus sind so vielgestaltig, dass es besser ist, den Rheumatologen einmal zu viel als zu wenig um Rat zu fragen.
- Machen Sie die Sonne zu Ihrem Feind: Schübe werden leicht von Sonnenstrahlen ausgelöst. Und wenn es nicht anders geht, verwenden Sie Sunblocker mit Lichtschutzfaktor 60 (sie werden nicht mehr von den Krankenkassen übernommen). Weitere Schubauslöser sind die Pille sowie psychische und physische Belastungen. Viele Patienten reagieren auch empfindlich auf Medikamente, z. B. auf Lokalanästhetika beim Zahnarzt.
Weiterführende Informationen
- www.dgkl.de Suchbegriff Lupus – Ärztliche Leitlinie zur Diagnostik und Therapie des Systemischen Lupus erythematodes.
- www.lupus-rheumanet.org – Lupus Erythematodes Selbsthilfegemeinschaft e. V., Wuppertal: Informative und sorgfältig verfasste Internetseite.
- M. Schneider: Lupus erythematodes. Steinkopff, 2004. Ausführlicher und aktueller fachärztlicher Ratgeber.
Was sind Kollagenosen?
Als Kollagenosen bezeichnet man in der Medizin entzündliche rheumatologische Erkrankungen, bei denen der Befall des Bindegewebes das auffälligste Kennzeichen ist.
Zur Gruppe der Kollagenosen im engeren Sinne gehören das Sjögren-Syndrom, die systemische Sklerodermie, die Poly-/Dermatomyositis, die Mischkollagenose, der systemische Lupus erythematodes und das Antiphospholipid-Syndrom.
Die Ausprägungen und Verläufe der Krankheiten sind unterschiedlich, teilweise treten auch Mischformen auf. Im Blut lassen sich häufig antinukleäre Antikörper nachweisen (Leitbefunde: ANA und ENA, extrahierbare nukleäre Antikörper).